{kind=link}
La investigación cardiovascular da un paso adelante en la comprensión y tratamiento de la miocardiopatía hipertrófica, la enfermedad genética del corazón más frecuente. Un equipo liderado por el Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), en colaboración con un grupo internacional de investigadores, ha identificado un nuevo mecanismo molecular implicado en el desarrollo de esta patología y ha confirmado que el mavacamten, el primer tratamiento dirigido disponible para esta enfermedad, resulta eficaz frente a diferentes tipos de mutaciones genéticas. Los resultados han sido publicados en la revista científica Nature Cardiovascular Research.
La relevancia del hallazgo es especialmente significativa debido a la elevada incidencia de esta enfermedad. La miocardiopatía hipertrófica constituye la enfermedad genética cardiovascular más frecuente y representa la causa más habitual de muerte súbita en personas jóvenes y deportistas. Según los datos epidemiológicos recogidos en la investigación, se estima que afecta aproximadamente a una de cada 250 a 500 personas en la población general. En España, alrededor de 95.230 personas conviven con esta patología.
La enfermedad se caracteriza por un engrosamiento anormal del músculo cardíaco y por una contractilidad excesiva del corazón. Esta hiperactividad del músculo cardíaco dificulta el bombeo normal de la sangre y, en los casos más graves, puede desencadenar arritmias potencialmente fatales. La patología está provocada por mutaciones en los genes responsables de codificar las proteínas del sarcómero, la estructura molecular encargada de la contracción cardíaca. Entre estos genes destaca especialmente MYBPC3, que codifica la proteína C de unión a la miosina cardíaca (cMyBP-C), una de las proteínas que con mayor frecuencia aparece implicada en esta enfermedad.
El estudio aporta ahora una nueva perspectiva sobre el funcionamiento de determinadas mutaciones de este gen que, hasta el momento, no habían sido completamente comprendidas. Según explica Laura Sen-Martín, autora principal de la investigación, «El trabajo se centra en estudiar el mecanismo molecular de un subgrupo de mutaciones en MYBPC3 que, a diferencia del tipo de mutación más habitual, no reducen la cantidad de la proteína, sino que alteran su capacidad de interactuar con otras proteínas del corazón. Hasta ahora, el mecanismo exacto por el que estas mutaciones causan la enfermedad no estaba bien definido».
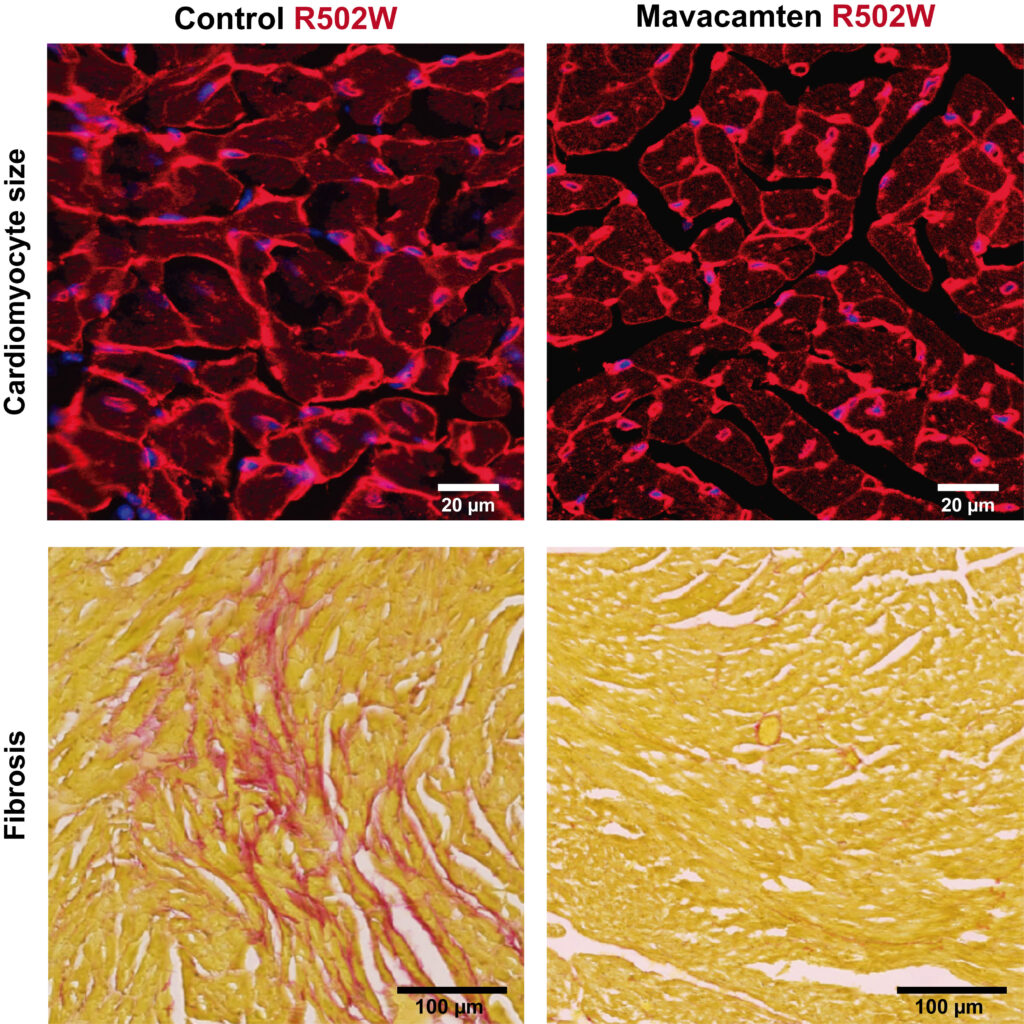
Para profundizar en este fenómeno, el equipo desarrolló un modelo experimental en ratón capaz de reproducir las principales características de la enfermedad mediante la variante genética R502W. El análisis de este modelo permitió comprobar que esta mutación disminuye la capacidad de la proteína cMyBP-C para interactuar con la miosina, considerada el motor molecular responsable de la contracción cardíaca. Según los autores, esta alteración en la interacción entre ambas proteínas constituye un nuevo mecanismo de patogenicidad para un subgrupo concreto de pacientes afectados por miocardiopatía hipertrófica.
Una vez identificado este nuevo mecanismo, los investigadores quisieron determinar si el mavacamten seguía siendo eficaz pese a que la mutación desencadena procesos moleculares diferentes a los observados en otras variantes de la enfermedad. El fármaco, que actúa directamente sobre la miosina modulando su actividad y reduciendo la contracción excesiva del corazón, es actualmente el único tratamiento dirigido disponible para la miocardiopatía hipertrófica.
Los resultados fueron positivos. El tratamiento consiguió frenar el remodelado patológico del músculo cardíaco tanto en el modelo experimental con la mutación R502W como en otro modelo caracterizado por la pérdida completa de la proteína cMyBP-C. Además, en el caso de los ratones portadores de la variante R502W, el tratamiento mejoró la tolerancia al ejercicio, un aspecto especialmente relevante para valorar la evolución funcional de la enfermedad.
La eficacia del medicamento también fue comprobada en tejido cardíaco generado en laboratorio a partir de cardiomiocitos humanos obtenidos mediante células madre inducidas. En este modelo experimental, el mavacamten logró reducir la fuerza excesiva de contracción característica del tejido enfermo, reforzando así la posible relevancia clínica de los resultados obtenidos por el equipo investigador.
El investigador principal del estudio, el doctor Jorge Alegre-Cebollada, líder del Grupo de Mecánica Molecular del Sistema Cardiovascular del CNIC e investigador del CIBERCV, destaca el impacto que ya está teniendo este tratamiento en la práctica clínica. En sus palabras, «El mavacamten y moléculas similares están transformando el tratamiento de pacientes con miocardiopatía hipertrófica; sin embargo, no todos los pacientes responden de la misma manera». Asimismo, añade que «este trabajo sugiere que la causa de esta efectividad desigual no se debe a las diferentes mutaciones que portan los pacientes».
La investigación concluye que el mavacamten mantiene su eficacia con independencia del mecanismo molecular responsable del desarrollo de la enfermedad, una conclusión que amplía potencialmente el número de pacientes que podrían beneficiarse de este tratamiento. Además, el nuevo modelo murino desarrollado durante el estudio se convierte en una herramienta de gran valor para evaluar futuras estrategias terapéuticas dirigidas específicamente a este subgrupo de pacientes.
En este sentido, Laura Sen-Martín apunta nuevas posibilidades de investigación al afirmar que «Por ejemplo, nuestro nuevo modelo experimental puede ser usado para entender si la administración temprana de mavacamten puede mejorar los resultados terapéuticos, un asunto que no está resuelto todavía en el ámbito clínico».
El trabajo ha contado con financiación principalmente del Ministerio de Ciencia, Innovación y Universidades, a través de diferentes proyectos de investigación, además del apoyo de la Comunidad de Madrid. Laura Sen-Martín desarrolla su labor investigadora gracias a una ayuda predoctoral financiada por el Ministerio.
El estudio, titulado Mavacamten shows broad benefit in human and mouse models of MYBPC3-related hypertrophic cardiomyopathy, ha sido publicado en Nature Cardiovascular Research y representa un nuevo avance en el conocimiento de la miocardiopatía hipertrófica, al identificar un mecanismo molecular hasta ahora no definido y confirmar que el principal tratamiento dirigido disponible mantiene su eficacia incluso cuando la enfermedad se desarrolla a través de mecanismos biológicos distintos. Este hallazgo abre nuevas perspectivas para ampliar las opciones terapéuticas y continuar investigando estrategias que permitan mejorar el abordaje de una enfermedad genética que afecta a decenas de miles de personas en España y constituye una de las principales causas de muerte súbita entre la población joven.